We use a multielectrode array to record the activity of thousands of neurons simultaneously.
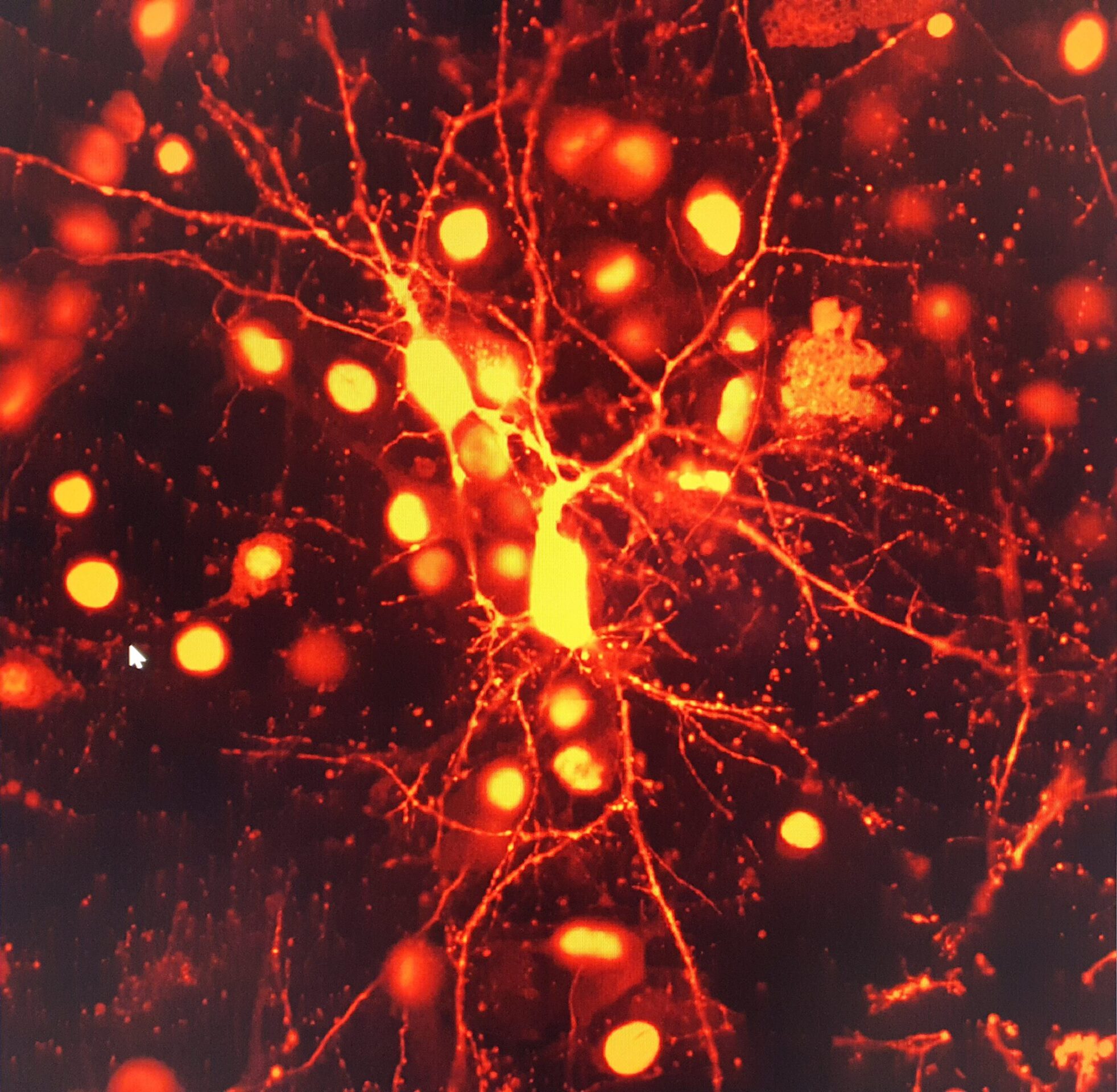
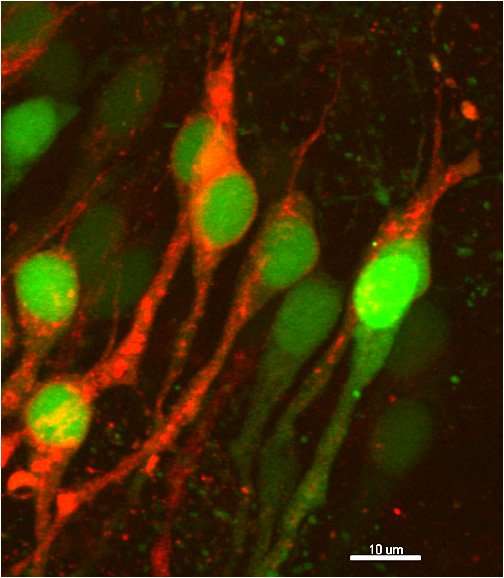
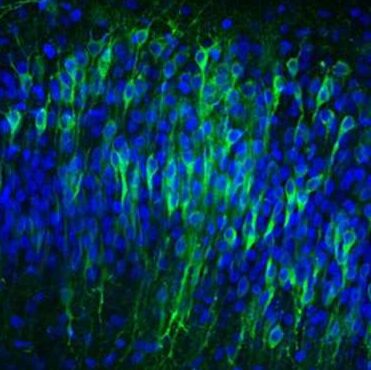
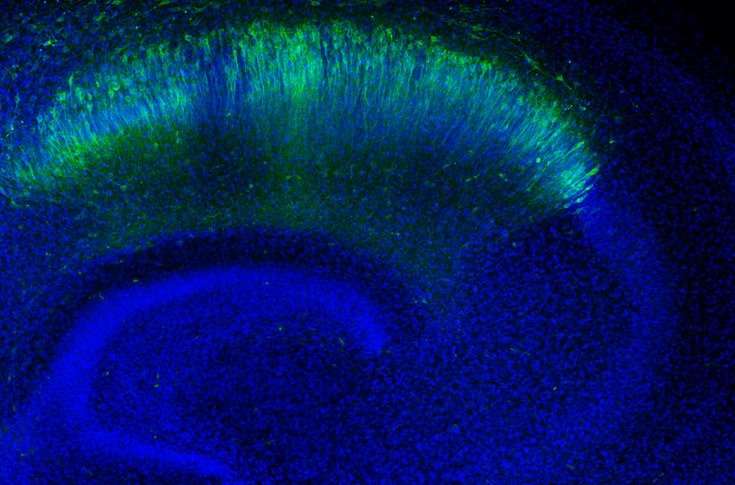
We use super-resolution confocal imaging and state-of-the-art software of image analysis, such as Imaris, Neurolucida and Neuroinfo.
Most of our studies involve a thorough behavioral analysis. For this purpose, our laboratory uses several tests that allow us to evaluate a wide range of rodent behaviors (Barnes Maze, Fear Conditioning, Novel Object Location, and more).

Tachykinin receptor 3 (TACR3) is a member of the tachykinin receptor family and falls within the rhodopsin subfamily. As a G protein-coupled receptor, it responds to neurokinin B (NKB), its high-affinity ligand. Dysfunctional TACR3 has been associated with pubertal failure and anxiety, yet the mechanisms underlying this remain unclear. Hence,...
Read Article
Synaptic impairment might precede neuronal degeneration in Parkinson’s disease. However, the intimate mechanisms altering synaptic function by the accumulation of presynaptic α-synuclein in striatal dopaminergic terminals before dopaminergic death occurs, have not been elucidated. Our aim is to unravel the sequence of synaptic functional and structural changes preceding the symptomatic...
Read Article
The capacity to learn new efficient systemic behavior is a fundamental issue of contemporary biology. We have recently observed, in a preliminary analysis, the emergence of conditioned behavior in some individual amoebae cells. In these experiments, cells were able to acquire new migratory patterns and remember them for long periods...
Read Article