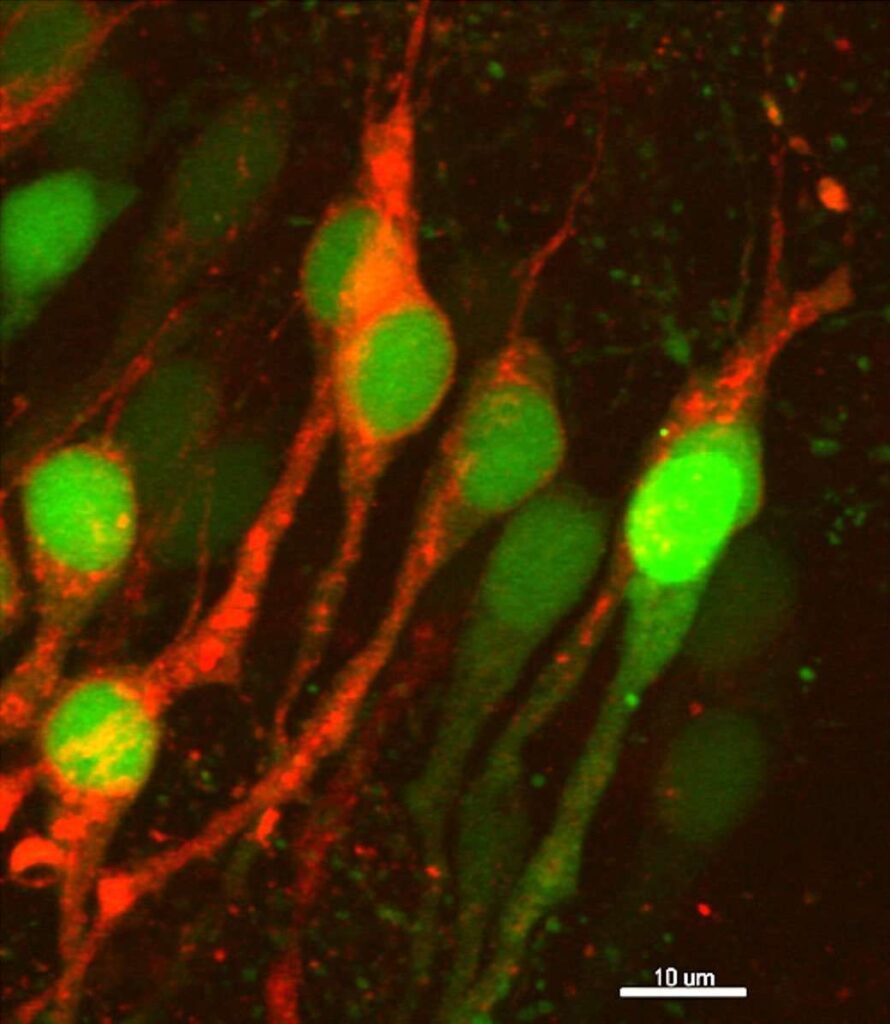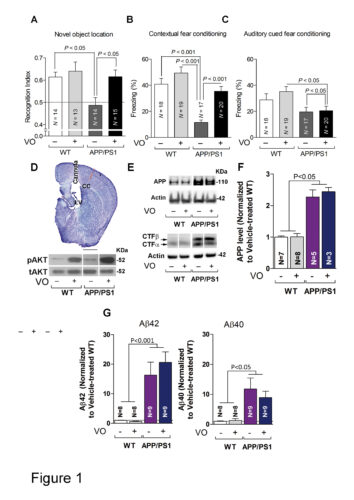
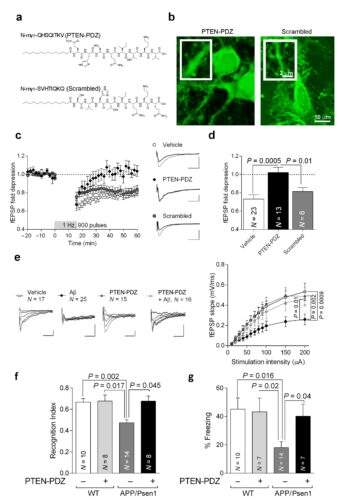
Dyshomeostasis of amyloid-β peptide (Aβ) is responsible for synaptic malfunctions leading to cognitive deficits ranging from mild impairment to full-blown dementia in Alzheimer's disease. Aβ appears to skew synaptic plasticity events toward depression. We found that inhibition of PTEN, a lipid phosphatase that is essential to long-term depression, rescued normal synaptic function and cognition in cellular and animal models of Alzheimer's disease. Conversely, transgenic mice that overexpressed PTEN displayed synaptic depression that mimicked and occluded Aβ-induced depression. Mechanistically, Aβ triggers a PDZ-dependent recruitment of PTEN into the postsynaptic compartment. Using a PTEN knock-in mouse lacking the PDZ motif, and a cell-permeable interfering peptide, we found that this mechanism is crucial for Aβ-induced synaptic toxicity and cognitive dysfunction. Our results provide fundamental information on the molecular mechanisms of Aβ-induced synaptic malfunction and may offer new mechanism-based therapeutic targets to counteract downstream Aβ signaling.